Esclerose Lateral Amiotrófica (ELA) ou Doença dos Neurônios Motores
 |
Resumo. A ELA é uma doença neurodegenerativa progressiva que afeta os neurônios motores superior (NMS) e inferior (NMI). O diagnóstico de certeza da ELA é feito pelo neurologista, em 4 etapas: clínica, eletroneuromiografia (ENMG), confirmação e exclusão. Para diagnóstico definido é preciso achados clínicos e de ENMG consistentes com NMS e NMI, em 3 regiões do corpo, acrescido da exclusão de (clínica, radiológica etc) de outras doenças. Em virtude de tratar-se de um diagnóstico complexo, nosso serviço reserva aos pacientes com ELA, sua família e seu (s) médico (s), o direito a debater e revisar conosco seu diagnóstico, tomando como base o texto e as referências aqui apresentadas, assim como aquelas que encontram-se no site da Associação Brasileira de ELA (ABRELA: https://www.abrela.org.br/).
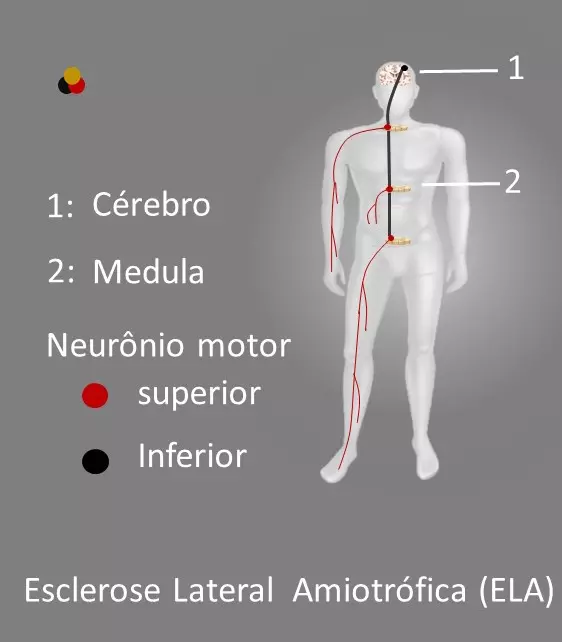
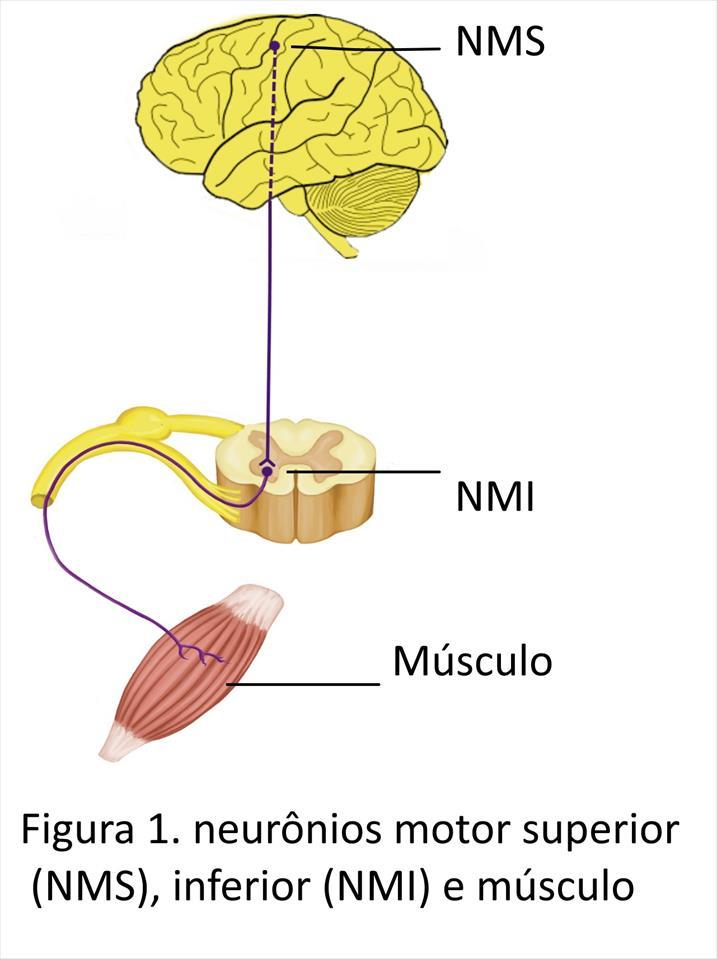
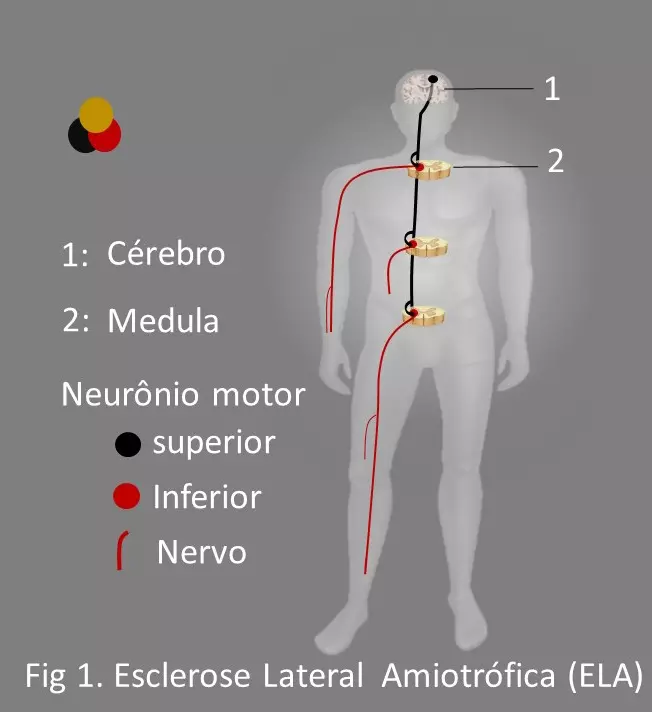
O que é a ELA e como é diagnosticada. A ELA é uma doença neurodegenerativa rara e seletiva dos NMS e NMI, afeta mais o sexo masculino (2M:1F), têm início dos sintomas após os 45 anos e aumenta sua incidência a cada década subsequente da vida. O termo doença dos neurônios motores (DNMs) origina-se de que a motricidade (força muscular) no ser humano, depende no sistema nervoso de dois grupos de neurônios motores (figura 1), um NMS que fica no córtex cerebral e daí se projeta para NMI, distribuídos difusamente na medula espinhal e tronco cerebral. Os NMI subsequentemente ramificam-se através dos nervos periféricos, fazendo a inervação dos músculos voluntários de todo o corpo.
 |
O resultado final das lesões do NMI e NMS é um quadro de atrofia muscular difusa e fraqueza, com perda importante de funções motoras básicas. Embora durante o ciclo de vida normal ocorra uma degeneração destes neurônios com o envelhecimento, na ELA esta degeneração é muito mais intensa, notadamente na sexta e sétima década de vida. Na atualidade o diagnóstico da ELA feito pelo neurologista, baseado-se nos critérios de Awaji-Shima (CAS, tabela 2), os quais vem substituíndo os critérios El Escorial revisados (CEEr, tabela 1). Os critérios são utilizados pois a ELA não têm um marcador (exame) característico, como ocorre com a glicemia no diabetes mellitus, a creatinina na insuficiência renal etc. O diagnóstico é feito em 4 etapas a saber: clínico, ENMG, confirmação e exclusão
Etapa 1: Diagnóstico clínico. O início clínico da ELA, em geral é assimétrico comprometendo um membro, posteriormente se generalizando para todos os membros, tronco e crânio, em geral mantendo um padrão assimétrico e multifocal. As principais manifestações de NMI e NMS são:
-
Disfunção de NMS: fraqueza, reflexos tendíneos vivos, presença de reflexos anormais (sinal de Babinski e outros)
-
Disfunção de NMI: fraqueza, fasciculações, atrofia, atonia
-
Disfunção dos neurônios motores do tronco cerebral: disfagia, disartria
Após computadas estas evidências de NMI e NMS clinicamente segue-se
 |
Etapa 2: Evidências de NMI pela ENMG. O exame em geral é feito em 4 a 6 regiões do corpo, mas caso o diagnóstico seja confirmado nas 3 primeiras regiões, o exame de mais uma região (a quarta) é mais que suficiente. Dessa forma a prudência mínima é examinar 4 regiões de 6 regiões previstas nos CAS, conforme abaixo (a numeração é do autor):
- Região 1: bulbar/cranial,
- Região 2 e 3: cervical direita e esquerda que inclui os 2 membros superiores,
- Região 3 e 4: torácica que inclui alguns músculos do abdome
- Região 5 e 6: lombosacra que inclui os 2 membros inferiores.
Na ENMG o diagnóstico é definido quando se evidencia desinervação ativa e crônica, em 3 das 6 regiões previstas dos CAS, sendo o diagnóstico provável quando em 2 regiões, e possível quando em apenas 1. Importante da ENMG é que ela pode mostrar desinervação tanto em regiões alteradas, quanto naquelas normais do ponto de vista clínico. Alterações devem ser demonstradas em pelo menos 2 músculos de diferentes raízes em cada membro superior (C5 e C7 por exemplo) e em cada membro inferior ( L4 e S1 por exemplo). Também os músculos alterados devem ser inervados por diferentes nervos, por exemplo os nervos axilar e radial em cada membro superior e os nervos femoral e tibial em cada membro inferior. Na região bulbar/cranial a exigência é de um único músculo alterado (trapézio ou língua), enquanto na região torácica a musculatura paraespinhal ou abdominal. A presença de fasciculação conta como desinervação, caso associada a potenciais de unidade motora neurogênicos. A ausência de fasciculação deve causar dúvida no diagnóstico embora não o exclua.
Etapa 3: Confirmação do diagnóstico. A partir das evidências clínicas e da ENMG, o neurologista confirma o diagnóstico em graus de evidência (certeza), conforme as definições do CAS abaixo:
- ELA clinicamente definida (típica): evidência clínica ou eletrofisiológica de NMS + NMI na região bulbar e pelo menos em duas regiões espinhais, ou; a presença de sinais NMS + NMI em 3 regiões espinhais.
- ELA clinicamente provável: evidência clínica ou eletrofisiológica de NMS + NMI em duas regiões e sinais de NMS rostralmente aos sinais de NMI
- ELA clinicamente possível: NMS + NMI em somente uma região, ou; NMS isoladamente em 2 ou mais regiões, ou; NMI rostralmente aos sinais de NMS. Estudos de neuroimagem e laboratoriais excluem outros diagnósticos
Etapa 4: diagnóstico de exclusão. A exclusão é sempre necessária mesmo quando na ELA foi definida em 3 regiões. Quando provável ou possível a decisão é disjuntiva: excluir outras doenças ficando com um diagnóstico provável de ELA, ou; excluir o diagnóstico de ELA em favor de outra doença. Neste caso as exclusões envolvem as manifestações de NMI, NMS, NMS e NMI, nem NMS nem NMI e bulbar.
NMS: Na espasticidade a RNM do crânio e coluna permite a exclusão de várias doenças notadamente mielopatia cervical, esclerose múltipla, tumor medular, siringomielia, malformação na junção crânio cervical, etc. Também importante relativamente a manifestações de NMS são esclerose lateral primária (ELP) e paraparesia espástica forma hereditária e tropical.
NMI: A síndrome pós pólio apresenta um quadro de NMI similar à ELA diferenciando-se pelo antecedente e uma desinervação mais crônica que aguda. Atrofia muscular progressiva (AMP), neuropatia motora tipo Charcot Marie Tooth e amiotrofia espinhal do adulto, são doenças com uma progressão mais lenta, maior sobrevida e desinervação menos intensa que a ELA. A AMP ficou conhecida em função de um portador famoso, o físico Stephen Hawking. Quando a apresentação clínica é multifocal com predomínio nos MMSS, deve ser considerado o diagnóstico de neuropatia motora multifocal, transtorno bastante cogitado no diagnóstico de exclusão da ELA, e que tratamos no capítulo específico. Atrofia em um único membro ou monomélica é rara. Fasciculacoes benignas é o termo reservado para o quadro de fasciculações, que ocorrem em várias regiões do corpo, mas sem evidências clínicas e de ENMG consistentes com NMI.
NMS e NMI: Manifestações de NMI (com ou sem NMS) também pode ocorrer nas doenças espinocerebelares hereditárias e quadros esporádicos tidos como atrofia multissistêmica, atrofia olivopontocerebelar e adrenomieloneuropatia, nas quais podem estar associados variavelmente um complexo de manifestações (degeneração sistêmica) incluindo NMI, NMS, polineuropatia sensitiva, ataxia etc. Um exemplo dessas doenças são a atrofia multissistêmica e as ataxias espinocerebelares incluindo nestas a doença de Joseph-Machado (ver doutorado).
Bulbar: Em caso de alterações na fala e na deglutição, é importante considerar que num pequeno percentual dos casos de ELA, a doença têm início na musculatura bulbar, depois se generalizando. Outros diagnósticos a se considerar neste caso são: a paralisia bulbar progressiva (PBP), a qual fica restrita a região do crânio, afetando músculos da face, mandíbula, faringe e laringe, e; mal formação da junção crânio vertebral, miastenia gravis, polimiosite e distrofia muscular.
Nem NMS nem NMI. Se existe déficit motor ou fadiga importante, e a clínica e a ENMG não sugerem NMI nem NMS, testes adicionais podem identificar, miastenia gravis, miopatias hereditárias, polimiosite, síndrome miastênica, hiperparatireoidismo, hipertireoidismo, miosite de inclusão, etc. No diabetes mellitus, por exemplo a caquexia e a radiculoplexopatia são muito frequentemente confundidas com a ELA em virtude do emagrecimento importante e déficit motor extenso, mas estas condições, diferenciam-se da ELA por apresentar um curso mais limitado e benigno, sem evidências de NMS.
Referências
Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000 Dec;1(5):293-9.
de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3): 497-503.
Kimura J. Disorders of spinal cord (Motor neuron Disease). In: Electrodiagnosis in diseases of nerve and muscle, principles and practice, 4rd ed (ebook kindle), New York: Oxford University Press (USA) 2013
Nimeshan Geevasinga, Clement T Loy, Parvathi Menon, et al. Awaji Criteria Improves the Diagnostic Sensitivity in Amyotrophic Lateral Sclerosis: A Systematic Review Using Individual Patient Data. Clin Neurophysiol 2016 Jul;127(7):2684-91.